|
Tertiary structure predictions of proteins:
Die Abfolge der Aminosäuren in der Polypeptidkette bestimmt unter definierten Umgebungsverhältnissen deren Faltung (oder Entfaltung) in eine festgelegte dreidimensionale Struktur, die Tertiärstruktur. Es gibt zwei Methoden, die 3D-Struktur eines Proteins experimentell in hoher Auflösung zu bestimmen, entweder per Röntgenkristallografie oder per NMR-Spektroskopie. Zur NMR-Methode finden sich gute Übersichtsbeiträge am Birkbeck-College (Kurt D. Berndt), am Imperial-College (Henry Rzepa) sowie an der Queens University. Schließlich sei noch auf die Methodenzusammenstellung auf der Webseite der PDB-Proteindatenbank hingewiesen.
Die Faltung eines Proteins beinhaltet so viele Wechselwirkungen zwischen den Atomen der Polypeptidkette - und der Umgebung (z.B. Wassermoleküle oder Lipide), dass seine 3D-Struktur (noch) nicht im voraus berechnet werden kann.
A good (but not very recent) introduction into the problem of structure prediction is available from Rob Russell at the EMBL in Heidelberg.
ab initio modeling:
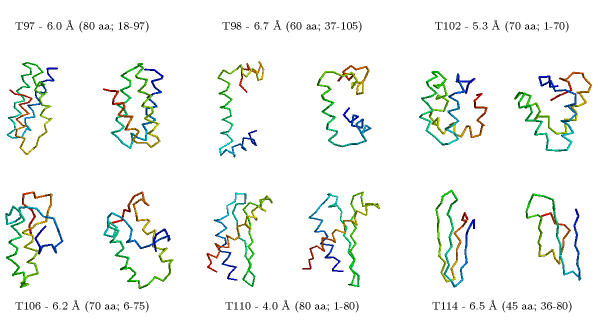
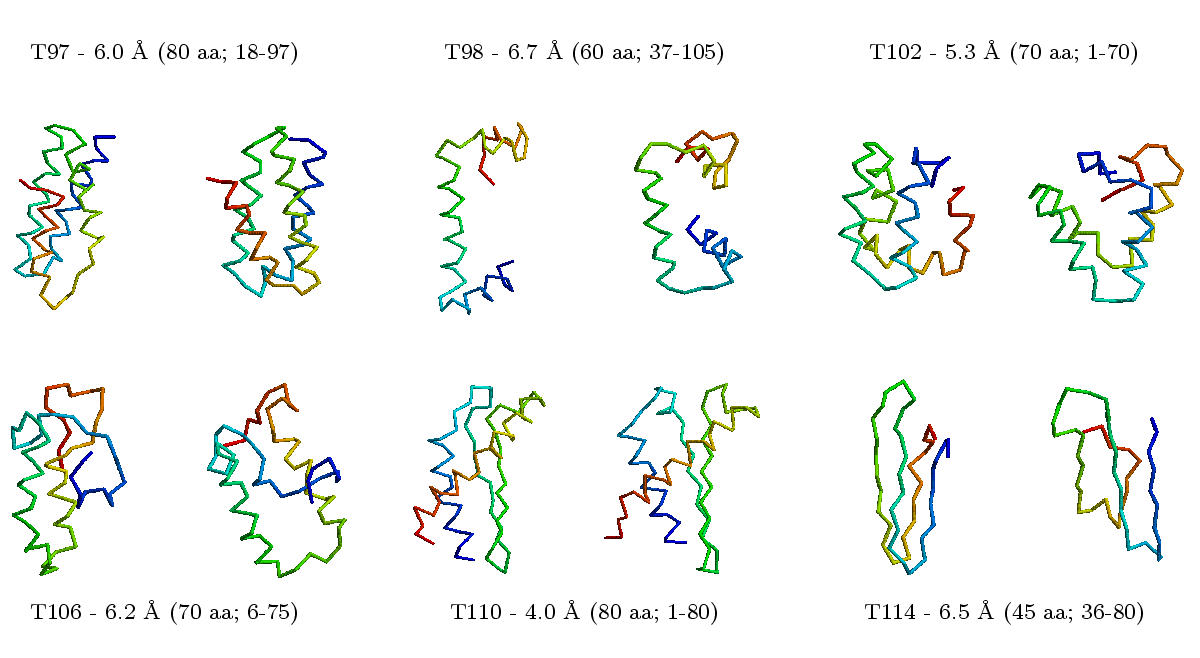
One possible approach is to test all theoretically possible bond angles between atoms and to calculate the stability of the resulting structures.
This approach requires too many calculations and cannot be completed within a reasonable period of time. Therefore, this approach is limited to short model peptides.
Alternatively, one can predict the conformation of short peptide fragments of the protein or extract possible conformations from known 3D structures.
In a second step, one can then combine these peptide fragments to a complete 3D structure ("fragment assembly").
Example (small, big).
- Protinfo - protein modelling server
- ROSETTA

- ROBETTA
Homology Modeling:
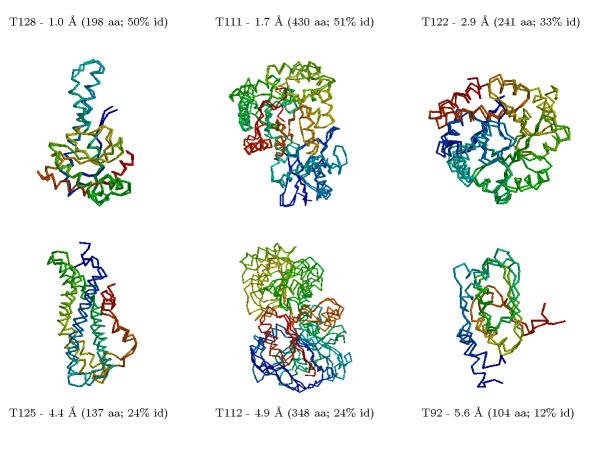
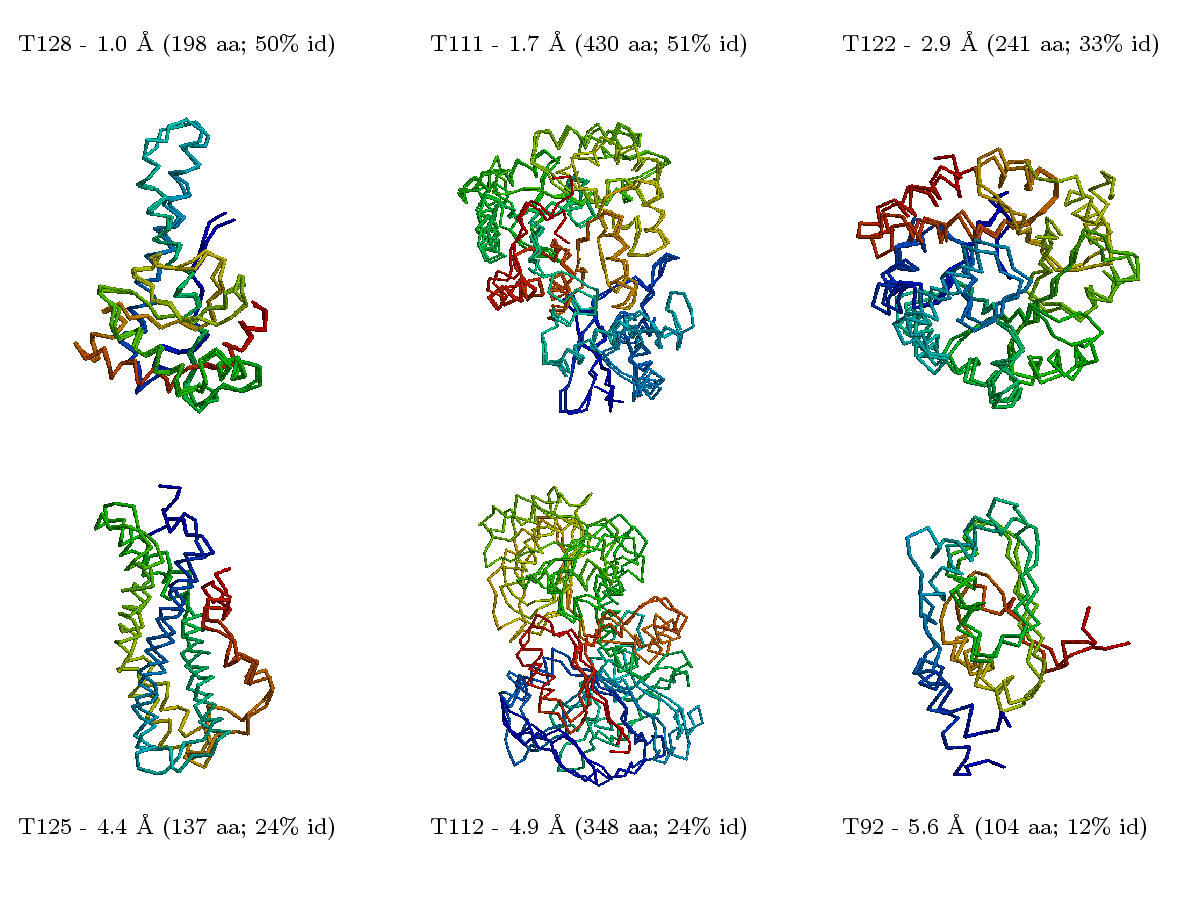
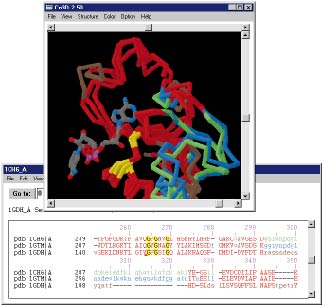
Aufgrund der Fortschritte in der Aufklärung von Genomsequenzen und 3D-Strukturen steigt die Chance, dass man zu einer Aminosäuresequenz ein homologes Protein finden wird, dessen Struktur bekannt ist. Wenn die Ähnlichkeit gross genug ist (> 30 %, besser > 50 % identische Aminosäuren), kann man versuchen, mittels Homology-Building für das unbekannte Protein ein Modell seiner Struktur zu berechnen. Dabei werden die Bereiche, die sich in einer definierten Sekundärstruktur befinden, am besten vorausberechnet werden, wohingegen oberflächen-exponierte Loops oder frei-bewegliche Seitenketten nur schlecht modelliert werden können. Beispiel (klein, groß).
Interessante Online-Kurse zum Molecular Modelling gibt es am Centre for Molecular and Biomolecular Informatics (CMBI) in Nijmegen, am Center for Biological Sequence Analysis (CBS) in Kopenhagen, und an der Mount Sinai School of Medicine in New York.
- SWISS-MODEL (GlaxoWellcome) ist ein PublicDomain-Server, der solche Strukturvorhersagen erlaubt.

- CPHmodels erlaubt ein Homology-Building mittels eines neuronalen Netzwerkes.
Weitere Server zum Homology Modelling sind:
- 3D-JIGSAW vom Cancer Research Institute (UK) modelliert nicht nur die main chain-Atome, sondern auch die side chain-Atome
- GENO3D am Institut de Biologie et Chimie des Protéines in Lyon
- SDSC1 am San Diego Supercomputer Center
- An der Rockefeller-University wurde das Programm MODELLER entwickelt; dabei handelt es sich allerdings nicht um einen Webserver. Weitere Software-Pakete zum Molecular Modelling sind WHAT IF sowie Insight II und Cerius2 von Accelrys Inc..
Threading:
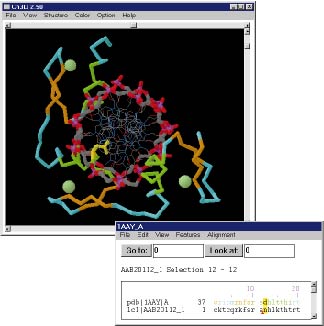
Ein alternativer Ansatz besteht darin, ausgehend von vorhandenen Protein-Folds zu versuchen, eine Sequenz in alle möglichen Folds "einzupassen". Diese Methode wird als Threading bezeichnet. Sie bezieht ihre Berechtigung aus der Tatsache, dass im Laufe der Evolution nur eine begrenzte Anzahl prinzipieller Protein-Folds entstanden ist (ca. 1000) und somit mit zunehmender Anzahl gelöster Strukturen die Erfolgschance dieses Ansatzes weiter steigt, da sämtliche Protein-Folds bzw -Superfamilien (ca. 15000) eines Tages in der Strukturdatenbank repräsentiert sein werden. Beispiel (klein, groß).
- Das Programm 3D-PSSM vom Imperial Cancer Research Institute ist auf einem Web-Server implementiert, der solch ein Threading im Internet erlaubt.

- Phyre (Protein Homology/analogY Recognition Engine) ist der Nachfolger von 3D-PSSM.
Weitere Server zum Threading bzw. zur Fold Recognition gibt es an folgenden Adressen:
- DOE Fold-Server (FRSVR) an der University of California at Los Angeles (Login)
- FFAS (Fold & Function Assignment System) am Burnham Institute, La Jolla, California (Login)
- FFAS03 am Burnham Institute, La Jolla, California
- Fugue an der University of Cambridge, UK
- INBGU an der Ben Gurion University, Israel
- LOOPP (Learning, Observing and Outputting Protein Patterns) an der Cornell-University in Ithaca, NY.
- Pmembr: A threading method for membrane proteins.
- PROSPECT (PROtein Structure Prediction and Evaluation Computer Toolkit) am Oak Ridge National Laboratory
- SAM (Sequence Alignment and Modeling System) an der University of California at San Francisco
- Sausage (Sequence-structure Alignment Using a Statistical Approach Guided by Experiment ) an der Australian National University Canberra (replaced by WURST )
- ssPsi an der Stockholm University (OUTDATED)
- Superfamily an der University of Cambridge, UK
- genTHREADER an der Brunel University, Uxbridge, UK
- TUNE1D (Threading Using Neural nEtwork with one-dimensional profile) am Medical Research Council, UK
- 123D+ am NCIFCRF (USA)
- Pcons was the first consensus server for fold recognition. It can be used at the Bioinformatics Laboratory (IIMCB) in Warshaw and at the Stockholm Bioinformatics Center (CBS). Pmodeller is an addition on top of Pcons. In Pmodeller all atom models are built, and also evaluated using ProQ. It has been shown in CASP5 that Pmodeller works better than Pcons. Currently Pmodeller (based on Pcons-2) and Pmodeller-4 (based on Pcons-4) is used.
- 3D-Jury von BioInfo war der erfolgreichste Metaserver während des CASP5-Wettbewerbs.
- LiveBench ist eine Plattform, die verschiedene Threading-Server miteinander vergleicht.
- ProCeryon ist ein Software-Paket zum Threading, das nicht im Netz zur Verfügung steht.
Visualisierung von 3D-Strukturen:
Zur Visualisierung von 3D-Strukturen gibt es verschiedene Programme, von denen die meisten lokal auf dem eigenen Computer installiert werden müssen (was im Rahmen dieses Kurses leider nicht möglich ist):
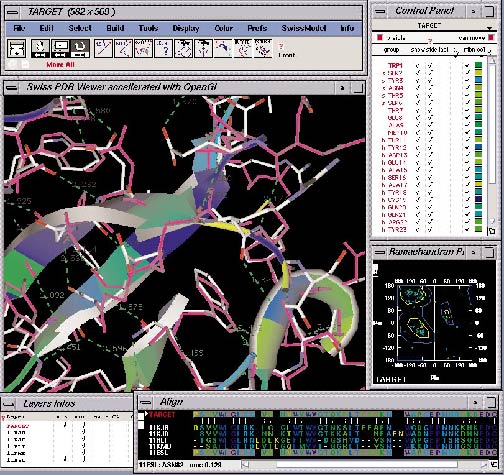
- Deep View (Swiss-Pdb Viewer), von Manuel C. Peitsch entworfen, wird ebenfalls von GlaxoWellcome bereitgestellt (Beispiel)

- Cn3D, am NIH bereitgestellt (Beispiel #1, Beispiel #2)

- RasMol, geschrieben von Roger Sayle, University of Massachusetts, Amherst, USA (Beispiel)

- RasMol and OpenRasMol
- MAGE und PREKIN, von Jane und David Richardson eingeführt, produzieren sog. Kinemages (kinetic images). Die Zeitschrift Protein Science erlaubt die Publikation solcher Abbildungen und stellt diese online zur Verfügung.
- Chime ist ein RasMol-basierendes Programm, das als Plug-In innerhalb eines Web-Browsers läuft und dort Strukturen darstellen kann, von denen die Koordinaten auf einer Webseite hinterlegt sind.
- Protein Explorer, Protein Morpher und Noncovalent Bond Finder (NCBF) sind Anwendungen, die Chime benötigen und in der Lage sind, 3D-Strukturen und in verschiedenen Ansichten darzustellen oder zu animieren bzw. Wasserstoffbrückenbindungen zu berechnen.
- Protein Explorer vs. RasMol
- WebMol, von Dirk Walther am EMLB Heidelberg geschrieben, ist ein JAVA-basierendes Programm, das die Visualisierung von 3D-Strukturen auf dem eigenen Webbrowser ermöglicht.

Desweiteren besteht die Möglichkeit, sich vorgefertigte Abbildungen von 3D-Strukturen anzuschauen oder als File (z.B. im PDF-Format) abzuspeichern:
- IMB Library
- Swiss-3DImage
- PDB-Images
- PDB-Molecule of the Month
Sequence examples:
- Database of amino acid sequences via Entrez
- 2-component response regulator HrpG from Xanthomonas campestris pv. vesicatoria
- 2-component response regulator OmpR from Escherichia coli
- 2-component response regulator PhoB from Escherichia coli
- AraC-type regulator HrpX from Xanthomonas campestris pv. vesicatoria
- Arabinose regulator AraC from Escherichia coli
- Multiple antibiotic resistance protein MarA from Escherichia coli
Latest update of content: September 20, 2005
Ralf Koebnik
Institut de recherche pour le dèveloppement
UMR 5096, CNRS-UP-IRD
911, Avenue Agropolis, BP 64501
34394 Montpellier, Cedex 5
FRANCE
Phone: +33 (0)4 67 41 62 28
Fax: +33 (0)4 67 41 61 81
Email: koebnik(at)gmx.de
Please replace (at) by @.
 Back to main page
Back to main page
|

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}